Klinische studies binnen het UZA
Wat is een klinische studie?
Een klinische studie betreft onderzoek bij gezonde vrijwilligers of patiënten met als doel de geneeskunde te verbeteren. Klinische studies trachten:
- nieuwe behandelingen voor bepaalde aandoeningen of ziektes te vinden en te testen (nog voordat deze op de markt worden gebracht);
- bestaande behandelingen te verbeteren;
- een beter inzicht in bepaalde aandoeningen en ziektes te verkrijgen (oorzaken, de impact van voeding, werkingsmechanismen en risicogroepen);
- ziektes beter op te sporen.
Een klinische studie bundelt dus wetenschappelijke vragen. Door antwoorden te zoeken op deze vragen, hopen onderzoekers de geneeskunde te verbeteren. Deelnemers aan klinische studies dragen bij aan de vooruitgang van de gezondheidszorg.
Alle klinische studies zijn streng gereglementeerd door de wet en worden ethisch goedgekeurd voordat ze van start gaan.
Commerciële versus academische klinische studies
Het initiatief voor een klinische studie kan uitgaan van het UZA, een andere niet-commerciële instelling, of een commerciële instelling zoals een farmaceutisch bedrijf. In de eerste twee gevallen spreken we over een academische studie, en in het laatste geval over een commerciële studie.
In het UZA worden zowel commerciële als niet-commerciële (academische) studies uitgevoerd. Voor de studiedeelnemers is er weinig verschil; ze worden in alle gevallen even goed beschermd en hebben dezelfde rechten en plichten. Dit wordt onder andere gewaarborgd door de internationale richtlijnen voor (goed) klinisch onderzoek (ICH-GCP).
Commerciële studies
Een ‘commerciële’ studie gaat meestal uit van een farmaceutisch bedrijf, die het ziekenhuis vergoedt voor de gemaakte onkosten.
De meeste onderzoeken naar nieuwe geneesmiddelen, medische hulpmiddelen of bijwerkingen zijn ‘commerciële’ studies. Vaak betreft het nationale of internationale multicentrische studies (uitgevoerd in verschillende ziekenhuizen of onderzoekscentra) met deelname van een grote groep patiënten.
Academische studies
Een ‘academische’ studie gaat uit van de onderzoekers zelf of een andere niet-commerciële organisatie, en wordt gefinancierd met eigen beperkte middelen, donaties of overheidsfondsen. Het onderzoek richt zich op geneesmiddelen en/of medische hulpmiddelen, vragenlijsten of beperkte interventies (een injectie, een RX-opname, …).
Academische studies worden vaak opgezet om meer te leren over een aandoening of om de geneeskundepraktijk te verbeteren.
(Niet-)interventioneel
Studies kunnen over het algemeen worden ingedeeld in interventionele en niet-interventionele studies. Deze indeling verduidelijkt het doel en de aanpak van het onderzoek.
Interventioneel
Een interventionele studie is een type onderzoek waarbij de onderzoekers actief ingrijpen in het leven of de behandeling van de deelnemers. Dit kan door bijvoorbeeld een bepaalde behandeling, therapie, medicatie of interventie toe te dienen om te kijken welk effect dit heeft op de gezondheid, het gedrag of een ander specifiek resultaat. Deze interventie kan op verschillende manieren gebeuren:
- een extra bloedafname
- het toedienen van een geneesmiddel in een andere dosis of formulering of voor een andere indicatie
- vragenlijsten
- een extra RX
- …
Bij een interventionele studie worden de deelnemers vaak willekeurig toegewezen aan verschillende groepen, zoals een experimentele groep die de interventie ontvangt en een controlegroep die een placebo of geen interventie krijgt. Dit maakt het mogelijk om causale relaties te onderzoeken, oftewel te kijken of de interventie daadwerkelijk het beoogde effect heeft.
Niet-interventioneel
Een niet-interventionele studie is een type onderzoek waarbij geen actieve interventie of behandeling door de onderzoekers wordt toegepast op de deelnemers. In plaats daarvan worden de deelnemers gewoon geobserveerd of gevolgd in hun natuurlijke omgeving, zonder dat er een gecontroleerde wijziging of manipulatie van hun gedrag, omstandigheden of behandeling plaatsvindt.
Het geneesmiddel wordt voorgeschreven onafhankelijk van de deelname van de deelnemer aan het onderzoek en als onderdeel van een therapeutische strategie (= Standard of Care (SoC)).
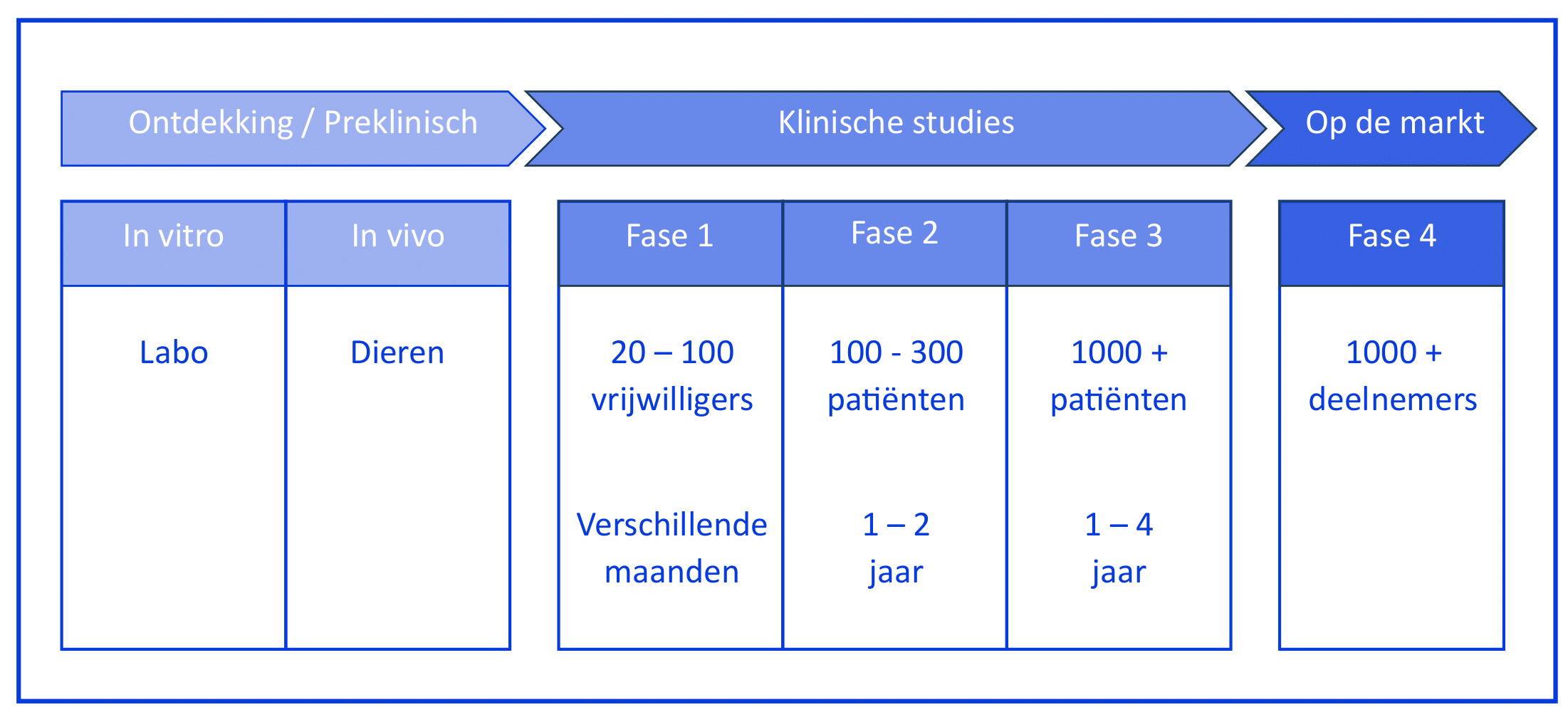
Klinische studie in vier fases
De ontwikkeling van een nieuw product of een nieuwe behandeling is een lang proces. Voordat er onderzoek op mensen plaatsvindt, wordt er uitgebreid preklinisch onderzoek gedaan, inclusief tests in het laboratorium en op proefdieren.

Deelnemen aan een klinische studie
Elk jaar melden veel mensen in het UZA zich vrijwillig aan voor klinische studies. Als er op de dienst waar je bent opgenomen een specifiek onderzoek loopt, kan het zijn dat je gevraagd wordt om hieraan mee te doen. Je bent volledig vrij om te beslissen of je wilt deelnemen. Als je interesse hebt om nu of in de toekomst betrokken te zijn bij klinische studies, kun je altijd terecht bij je behandelend arts voor meer informatie.
Ethische goedkeuring
Elke studie moet voor aanvang goedgekeurd worden door een erkend ethisch comité, zoals het Ethisch Comité UZA/UAntwerpen. Voor onderzoek naar nieuwe geneesmiddelen en medische hulpmiddelen is ook de goedkeuring van de Belgische autoriteit Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten (FAGG) nodig. Zo wordt gegarandeerd dat het onderzoek wetenschappelijk relevant en voor de deelnemers verantwoord is.
Ethisch Comité
Het Ethisch Comité van het UZA/UAntwerpen, kortweg het "Ethisch Comité", bestaat uit een diverse groep van personen met elk een eigen expertise, zoals artsen, verpleegkundigen, kinesitherapeuten, een jurist, een ethicus, een patiëntvertegenwoordiger, een apotheker, een psycholoog, een farmacoloog, een onderzoeksmethodoloog, een huisarts, een pediater en ook een aantal wetenschappers van de UAntwerpen. De samenstelling is conform de geldende wetgeving.Op regelmatige basis beoordeelt de vergadering alle wetenschappelijke onderzoeken in het UZA op nut, deelnamerisico’s, duidelijkheid van de informatie voor de deelnemers en vrije toestemming van de patiënt, alsook de omgang met patiëntgegevens. Bij bezwaren stelt het Ethisch Comité extra vragen aan de onderzoeker. Daarna geeft het Ethisch Comité een gunstig (goedkeuring) of een ongunstig (afwijzing) advies. Enkel na goedkeuring kan de studie starten. Het Ethisch Comité blijft de studie opvolgen, en kan ze indien nodig stopzetten, bv. omwille van onaanvaardbare bijwerkingen. Het Ethisch Comité waakt dus gedurende de hele duur van de studie over de belangen van de deelnemers.
Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten (FAGG)
In België staat het Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten (FAGG) als bevoegde overheid in voor het verzekeren van de kwaliteit en veiligheid van geneesmiddelen en gezondheidsproducten, voor zowel menselijk als diergeneeskundig gebruik, in ontwikkeling en op de markt.Voor onderzoek naar nieuwe geneesmiddelen en medische hulpmiddelen (interventionele studie) is de goedkeuring van het Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten (FAGG) nodig. Dit is niet vereist voor niet-interventionele studies.
Verloop van de studie
Protocol: leidraad van de studie
Na goedkeuring door het Ethisch Comité en zo nodig de Belgische autoriteit Federaal Agentschap voor Geneesmiddelen en Gezondheidsproducten (FAGG) kan de studie starten.
Het verloop gaat steeds volgens een protocol: een door de onderzoekers vastgelegd plan met daarin het doel van de studie, de selectiecriteria voor de deelnemers, alle onderzoeksstappen, mogelijke bijwerkingen, enz. De onderzoekers moeten dit protocol nauwgezet volgen, het is de leidraad van de studie.Selectie
De deelnamecriteria in het protocol bepalen welke deelnemers of patiënten toegelaten (inclusiecriteria) of uitgesloten (exclusiecriteria) worden.
Enkele vaak voorkomende exclusiecriteria zijn:
• leeftijd
• medische voorgeschiedenis
• zwangerschap of de kans daarop
• gelijktijdige deelname aan een andere klinische studie
• verslavingsgedragJouw rechten & plichten
Vrijwillige deelname
Deelnemen aan een klinische studie gebeurt altijd op vrijwillige basis. Je bent dus vrij om deel te nemen of niet. Jouw weigering heeft geen nadelige gevolgen voor de verdere relatie met jouw arts, de verpleegkundigen of het ziekenhuis: je zal altijd op de best mogelijke manier behandeld en verzorgd worden.
Recht op informatie en ‘informed consent’
Om een vrije en weloverwogen keuze te kunnen maken om al dan niet aan een studie deel te nemen, zal de onderzoeker jou uitgebreid informeren over onder andere het doel, het verloop van de studie en de eventuele bijwerkingen of risico’s.Van de arts of onderzoeker krijg je een informatiedocument en een toestemmingsformulier. Door dit te ondertekenen bevestig je dat je voldoende werd ingelicht, en geef je schriftelijk jouw geïnformeerde toestemming of ‘informed consent’ tot deelname.
Ook nadat je jouw toestemming verleende kan je op elk moment van de studie aan de onderzoeker, de arts of de studieverpleegkundigen meer informatie vragen.
Voor personen die hun toestemming niet kunnen geven (bv. minderjarigen, ...) kan in bepaalde situaties de toestemming gegeven worden door een wettelijke vertegenwoordiger. Het Ethisch Comité zal hiervoor zijn uitdrukkelijke goedkeuring verlenen.
Terugtrekkingsrecht
Je kan u op elk moment uit de studie terugtrekken zonder nadelige gevolgen voor jouw verdere behandeling of verzorging in het ziekenhuis. In het belang van je gezondheid moet een behandeling met geneesmiddelen soms eerst afgebouwd worden, of kan een controleonderzoek noodzakelijk zijn.
Vertrouwelijkheid van de gegevens
Alle studiegegevens worden strikt vertrouwelijk behandeld, conform de in België geldende privacy wetgeving. In publicaties van de studieresultaten blijft de identiteit van de deelnemers geheim.
Plichten
Naast rechten heb je als deelnemer ook enkele plichten: je moet stipt het behandelings- schema volgen, afspraken nakomen, en de voorgeschreven geneesmiddelen correct innemen. Wanneer je de afspraken niet nakomt, kan je uit de studie gezet worden.
Het is belangrijk jouw arts op de hoogte te brengen van alle bijwerkingen die je ervaart, als de bijwerkingen verergeren of als er iets verandert aan jouw medische situatie. Ook indien je opgenomen wordt in een andere ziekenhuis moet je dit zo snel mogelijk melden aan jouw arts.Vergoeding
Als je een vergoeding krijgt voor je deelname aan een studie, zal dit uitdrukkelijk vermeld staan in het Informed consent formulier (ICF) dat je vooraf ontvangt.
(wettelijke) bescherming
Gezonde deelnemers en patiënten die deelnemen aan klinische studies worden op verschillende manieren beschermd, zowel op Europees als op nationaal niveau. In België worden klinische studies geregeld door de wet van 7 mei 2004 (de ‘experimentenwet’), en de wet van 7 mei 2017 (geneesmiddelen) en de wet van 20 oktober 2020 (medische hulpmiddelen), en hun respectievelijke uitvoeringsbesluiten. In deze wetten staat onder meer de voorwaarden waaraan een experiment moet voldoen, de goedkeuringsprocedure, de verzekeringsverplichting enz.
Aansprakelijkheid & verzekering
Elke klinische studie is zo opgezet dat de risico’s voor de deelnemer zo klein mogelijk zijn. Helaas sluit dit niet uit dat er toch onverwachte problemen kunnen optreden. Daarom wordt er voor de deelnemers een bijkomende ‘foutloze’ verzekering afgesloten door de sponsor conform met de wetgeving. Dit impliceert dat schade veroorzaakt door de klinische studie wordt vergoed, ook als er geen duidelijke fout van de arts is aangetoond.
AansprakelijkheidDe opdrachtgever (initiatiefnemer) van een klinische studie is ‘foutloos’ aansprakelijk voor de schade die een deelnemer oploopt en die een rechtstreeks of onrechtstreeks verband met de studie vertoont. U als deelnemer moet dus enkel aantonen dat eventuele schade in verband staat met de studie. Je moet geen fout van de arts of onderzoeker aantonen.
VerzekeringDe opdrachtgever van een studie is verplicht zich voor dit risico te verzekeren. Academische studies waarvoor UZA verantwoordelijk is, worden gedekt door een algemene aansprakelijkheidsverzekering die het UZA heeft afsloten. Andere studies worden verzekerd door de farmaceutische firma of de instelling die de opdrachtgever is van de studie, bijvoorbeeld Universiteit Antwerpen. Het Ethisch Comité kijkt na of de verzekering in orde is.
"Compassionate use" & "medical need"
In bepaalde gevallen kan een patiënt een nog niet beschikbaar geneesmiddel krijgen (‘compassionate use’). Het geneesmiddel moet al wel een testprocedure hebben doorlopen. Soms krijgt een patiënt een geneesmiddel dat al wel vergund is, voor een andere indicatie dan waarvoor het vergund werd (‘medical need’).
Hierop is de regelgeving voor experimenten op mensen niet van toepassing. Hier gelden andere regels ter bescherming van de patiënt. Het kan toegepast worden bij patiënten met een ziekte die:
- chronisch, zwaar of levensbedreigend is
- niet met een beschikbaar of vergund geneesmiddel kan worden behandeld
Compassionate Use en Medical Need moeten ook voorgelegd worden aan het Ethisch Comité en de Belgische autoriteit (indien van toepassing).
General Data Protection Regulation (GDPR)
Alle studiegegevens worden strikt vertrouwelijk behandeld, volgens de Belgische privacywetgeving. Jouw identiteit blijft altijd geheim in publicaties over de studieresultaten.
Gezonde vrijwilligers
Veel klinische studies zijn afhankelijk van de deelname van mensen zonder gezondheidsproblemen. Deze "gezonde vrijwilligers" spelen een essentiële rol bij het verzamelen van waardevolle gegevens. Hun informatie dient als referentiepunt en wordt vergeleken met gegevens van mensen met specifieke aandoeningen of gezondheidsproblemen. Door hun bijdrage helpen gezonde vrijwilligers onderzoekers om beter inzicht te krijgen in ziekten en de effectiviteit van nieuwe behandelingen te beoordelen.
Patiëntenadviesraad
Met wetenschappelijk onderzoek boeken we vooruitgang in de geneeskunde en gezondheidszorg van morgen. Het UZA streeft ernaar patiënten en patiëntvertegenwoordigers te betrekken als volwaardige partners in haar wetenschappelijk onderzoek. Wie belang heeft bij zorg en onderzoek naar die zorg, moet zijn/haar stem kunnen laten horen. Daarom lanceerde het UZA begin 2023 de Patiëntenadviesraad voor Wetenschappelijk Onderzoek.
Contact
Clinical Trial Center (CTC)
Veelgestelde vragen
Wat is een klinische studie?
Een klinische studie is onderzoek dat bedoeld is om de medische kennis over de behandeling van ziekten of aandoeningen te vergroten. In klinische studies worden vrijwilligers (ook deelnemers genoemd) gerekruteerd zodat onderzoekers een experimenteel geneesmiddel kunnen testen en eventueel kunnen vergelijken met een standaardgeneesmiddel dat al beschikbaar.
Waarom zouden deelnemers meedoen aan een studie?
Een klinische studie vormt de basis voor de ontwikkeling van nieuwe geneesmiddelen. De resultaten van deze studies kunnen een verschil maken in de zorg voor toekomstige patiënten door informatie te verschaffen over de voordelen en risico’s van therapeutische geneesmiddelen. De huidige behandelingen voor ziekten zijn enkel beschikbaar doordat mensen zoals u meedoen aan een onderzoeksstudie.
Wat is een experimenteel geneesmiddel?
Een experimenteel geneesmiddel (of studiegeneesmiddel) is een geneesmiddel dat in een klinische studie wordt onderzocht. Experimentele geneesmiddelen kunnen al dan niet worden goedgekeurd door de nationale gezondheidsinstantie na onderzoek en op de markt gebracht worden. Elk experimenteel geneesmiddel moet echter worden beoordeeld, goedgekeurd en gecontroleerd door een ethisch comité om te kunnen worden getest op deelnemers aan een klinische studie.